This is a write-up of a talk I gave at the Boston Python Meetup on 23 July 2019, while I was Director of Bioinformatics at seqWell. It walks through three questions:
- What’s integer programming?
- What’s a gene library?
- How can we design a gene library using integer programming in Python?
What is integer programming?
Integer programming is optimization where the solution is required to be integer-valued. It’s a special case of linear programming: you maximize (or minimize) a linear objective function subject to a set of linear constraints, with the added restriction that the variables must take integer values.
It shows up all over the place — operations research, industrial production, scheduling — anywhere you’re allocating discrete, indivisible units rather than continuous quantities.
A small two-variable example. Maximize x₂ subject to:
max x₂
-x₁ + x₂ ≤ 1
3x₁ + 2x₂ ≤ 12
2x₁ + 3x₂ ≤ 12
x₁, x₂ ≥ 0
x₁, x₂ ∈ ℤWe’ll come back to this and solve it in Python below.
What is a gene library?
DNA synthesis is the “write” of molecular biology — the technology that lets us manufacture DNA sequences to order.
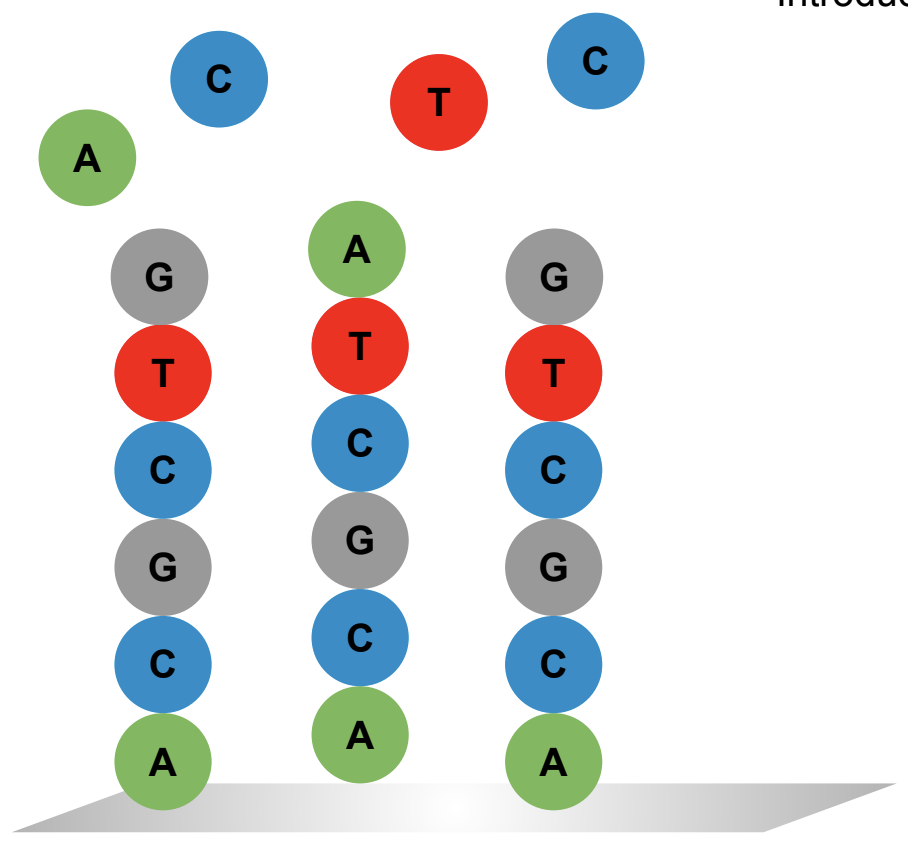
At each cycle of synthesis you add a nucleotide. If instead of a single nucleotide you add a mixture of nucleotides at a given cycle, you generate a diverse set of products: a gene library.
Why would we want that? Because of the central dogma — DNA is transcribed to RNA and translated to protein — and the genetic code maps three-base codons to amino acids. A library of DNA sequences becomes a library of proteins, which is exactly what you want when you’re screening for protein variants.
Degenerate nucleotides
These mixed positions are described with degenerate (IUPAC) nucleotide codes — letters other than A, C, G, T that each stand for a set of bases:
| Code | Bases | Code | Bases |
|---|---|---|---|
| B | C, G, T | R | A, G |
| D | A, G, T | S | C, G |
| H | A, C, T | V | A, C, G |
| K | G, T | W | A, T |
| M | A, C | Y | C, T |
| N | A, C, G, T |
So a codon like TWC (where W = A or T) encodes either TAC (Tyrosine) or TTC (Phenylalanine) — one written codon, two possible amino acids in the resulting library.
A single IUPAC codon can encode several amino acids at defined frequencies. For example SKR (S = C,G; K = G,T; R = A,G) expands to eight DNA codons that translate to a 25/25/25/25 mix of Arginine, Leucine, Glycine, and Valine.
The optimization problem
Here’s the design problem. Generate a gene library where:
- each sequence is 150 nucleotide bases long,
- the translation results in a target distribution of amino acids, and
- all sequences terminate in DNA base
C.
The target amino acid distribution (as percentages) might look like: Serine 20, Threonine 20, Alanine 9, Glycine 7, Aspartic acid 6, Glutamic acid 5, Isoleucine 5, Lysine 5, Leucine 5, Valine 5, Proline 4, Glutamine 4, Arginine 2, Tyrosine 2, Methionine 1, and 0 for the rest.
The key insight is that each degenerate codon contributes a known vector of amino acid frequencies. Picking how many of each IUPAC codon to use, so that the totals match the target distribution, is a system of linear equations with an integer solution — an integer program.
Solving it with Python
Two Python libraries can model integer programs: PuLP and Pyomo. Both can hand the model off to external solvers (CPLEX, COIN-OR CLP, GLPK, the commercial Gurobi and Xpress), and PuLP also ships with its own solver, so it works out of the box.
Warm-up: the small example in PuLP
First the two-variable example from above. We can write the constraints as a matrix X and a right-hand-side vector Y:
X = [[-1, 1],
[ 3, 2],
[ 2, 3]]
Y = [1, 12, 12]Create the integer variables:
import pulp
# create variables for x_1 and x_2
variables = [pulp.LpVariable('x_1', cat='Integer', lowBound=0),
pulp.LpVariable('x_2', cat='Integer', lowBound=0)]Initialize a maximization problem, add the constraints, then set the objective:
# initialize maximization problem
exampleLP = pulp.LpProblem('example', sense=pulp.LpMaximize)
# add constraints to LpProblem
for lhs, rhs in zip(X, Y):
exampleLP += lhs[0]*variables[0] + lhs[1]*variables[1] <= rhs
# add objective to LpProblem (maximize x_2)
exampleLP += variables[1]Solve and read out the solution:
# solve problem
status = exampleLP.solve()
assert pulp.LpStatus[status] == 'Optimal'
# get solutions
solutions = {'x_1': variables[0].value(),
'x_2': variables[1].value()}
# {'x_1': 1.0, 'x_2': 2.0}Designing the gene library
Now the real problem. Start with the imports and define the IUPAC nucleotide mapping:
from collections import Counter
import itertools
import pandas as pd
import pulp
# define IUPAC nucleotides
IUPAC = {'A': 'A',
'B': 'CGT',
'C': 'C',
'D': 'AGT',
'G': 'G',
'H': 'ACT',
'K': 'GT',
'M': 'AC',
'N': 'AGCT',
'R': 'AG',
'S': 'CG',
'T': 'T',
'V': 'ACG',
'W': 'AT',
'Y': 'CT'}Enumerate every possible IUPAC codon, and build the standard codon table:
# get all possible IUPAC codons
IUPAC_codons = [''.join(i) for i in itertools.product(IUPAC, repeat=3)]
# define codon table
trinucleotides = [''.join(i) for i in itertools.product('TCAG', repeat=3)]
amino_acids = 'FFLLSSSSYY**CC*WLLLLPPPPHHQQRRRRIIIMTTTTNNKKSSRRVVVVAAAADDEEGGGG'
codon_table = dict(zip(trinucleotides, amino_acids))For any IUPAC codon, expand it to its constituent DNA codons and count the amino acid frequencies it produces:
def process_codons(codon, IUPAC, codon_table):
# For a given IUPAC codon, return amino acids with frequencies
codons = [''.join(i) for i in
itertools.product(IUPAC[codon[0]], IUPAC[codon[1]], IUPAC[codon[2]])]
AAs = Counter([codon_table[c] for c in codons])
AA_dict = {AA: AAs[AA] / float(sum(AAs.values())) for AA in AAs}
return AA_dict
assert process_codons('SKR', IUPAC, codon_table) == \
{'G': 0.25, 'L': 0.25, 'R': 0.25, 'V': 0.25}Build the left-hand side of the system — a DataFrame of every IUPAC codon and its amino acid frequencies — plus a row that constrains the total number of codons (here, 50). The right-hand side Y is the target count of each amino acid:
# get dataframe of all codons with their amino acid frequencies
X = {c: process_codons(c, IUPAC, codon_table) for c in IUPAC_codons}
X = pd.DataFrame(X)
X = X.fillna(0)
# define total number of codons (50 total)
codon_sum = [[1]*len(X.columns)]
codon_sum = pd.DataFrame(codon_sum, index=['sum'], columns=X.columns)
X = X.append(codon_sum)
# define RHS of equation - counts of each amino acid
Y = [0.0, 4.5, 0.0, 3.0, 2.5, 0.0, 3.5, 0.0, 2.5, 2.5, 2.5, 0.5,
0.0, 2.0, 2.0, 1.0, 10.0, 10.0, 2.5, 0.0, 1.0, 50.0]Initialize the problem with one integer variable per IUPAC codon (how many of that codon to include), and add one equality constraint per amino acid:
# initialize problem
stem_problem = pulp.LpProblem('stems', sense=1)
# add variable for every IUPAC codon
variables = [pulp.LpVariable(x, lowBound=0, cat='Integer') for x in X.columns]
# add constraints to LpProblem
for row, y in zip(X.values.tolist(), Y):
stem_problem += sum([row[i]*variables[i] for i in range(len(row))]) == ySolve, and pull out the codons with non-zero counts:
# solve problem
status = stem_problem.solve()
assert pulp.LpStatus[status] == 'Optimal'
# get solutions
solutions = {v: v.value() for v in variables if v.value() != 0}
assert sum(solutions.values()) == 50The output is a recipe — the set of degenerate codons, and how many of each, that together produce the target amino acid distribution:
{KAC: 2.0, GWG: 1.0, GWT: 3.0, AKY: 4.0, ASB: 6.0,
AMA: 2.0, GAV: 1.0, GMV: 1.0, CAR: 1.0, AWR: 1.0,
RYK: 2.0, TYR: 5.0, VAA: 3.0, RSC: 14.0, MCA: 4.0}That’s the whole pipeline: a protein-design specification, expressed as a system of linear equations, solved as an integer program in a few lines of PuLP.
Thanks for reading. Questions welcome at jessicasmithbioinformatics@gmail.com.
Resources: